 |
|
Case Report
| ||||||
| Amyopathic Dermatomyositis (Anti-MDA 5 disease) with pulmonary fibrosis and features of lupus, without evidence of malignancy: A Case Report | ||||||
| Luis W. Dominguez1, Marissa Sansone1, Valentin Marian2 | ||||||
|
1Departments of Medicine, Jersey City Medical Center, 355 Grand Street, Jersey City, New Jersey, US 2Departments of Rheumatology, Jersey City Medical Center, 355 Grand Street, Jersey City, New Jersey, US | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article] [Similar articles in PubMed] [Similar articles in Google Scholar] |


| How to cite this article |
| Dominguez LW, Sansone M, Marian V. Amyopathic Dermatomyositis (Anti-MDA 5 disease) with pulmonary fibrosis and features of lupus, without evidence of malignancy: A Case Report. J Case Rep Images Orthop Rheum 2018;3:100014Z14LD2018. |
|
ABSTRACT
| ||||||
|
Introduction: Amyopathic Dermatomyositis (ADM) is a distinct subclass of Dermatomyositis (DM). It presents with similar skin findings of Gottron’s papules and cutaneous ulcerations, however it does not have myositis. Like DM, it is highly associated with malignancy, but unlike DM, there is a higher rate of progressive pulmonary fibrosis/interstitial lung disease (ILD). The presence of melanoma differentiation-associated gene 5 (MDA-5) antibodies are correlated with cutaneous findings but are not necessary for diagnosis. There is significant clinical and serological overlap between ADM and other autoimmune rheumatological conditions. Case Report: A 44-year-old Hispanic female with a medical history of hypothyroidism presented with an intermittent rash for the past six years. The rash was painful, pruritus and photosensitive. Physical exam revealed hyperpigmentation and keratosis over the metacarpal phalanges and periorbital erythema and edema. There were areas of alopecia and diffuse joint swelling and tenderness. Strength was intact. Pulmonary auscultation revealed diffuse crackles. Computed tomography of her chest confirmed pulmonary fibrosis. DM, complicated by a connective tissue disease was considered, however creatinine phospho-kinase (CPK) and aldolase levels were within normal limits. Accordingly, ADM was diagnosed and treatment was administered. MDA-5 antibodies were not available at any regional testing sites. Serology was positive for anti-nuclear (ANA) and cardiolipin antibodies. She failed azathioprine therapy, but responded to cyclophosphamide induction with bridging to mycophenolate, and Nintedanib for pulmonary fibrosis. Conclusion: ADM has a higher incidence of rapidly progressive pulmonary fibrosis, and significant overlap with other rheumatological conditions such as Lupus (SLE) and auto-immune hepatitis, as seen here. First line treatment is prednisone with steroid sparing agent azathioprine. Our patient failed first line treatment, but did respond to cyclophosphamide induction and mycophenolate maintenance, alongside Nintedanib for her pulmonary fibrosis.. Keywords: Amyopathic Dematomyositis, Anti MDA-5 disease, Dematomyositis overlap syndromes | ||||||
|
INTRODUCTION
| ||||||
|
Amyopathic Dermatomyositis (ADM) also known as dermatomyositis sine myositis, is a distinct subclass of dermatomyositis, distinguished by the classic findings of palmar papules, cutaneous ulcerations, nail fold erythema, and rapidly progressive ILD but without muscle weakness [1],[2]. Sontheimer (1999) suggested considering myositis as a spectrum of muscle and skin disease, with polymyositis on one end (being primarily muscular) to ADM (being primarily cutaneous) on the other, and DM in the middle (with both muscular and cutaneous manifestations) [3]. Diagnostic criteria for ADM was proposed to consist of a skin biopsyto rule out other etiologies, and the absence of muscle weakness or muscle enzyme elevation for at least 2 years [3].Of note, the average duration of cutaneous disease in ADM isaround 3.7 years [2]. This is likely secondary to a delay in diagnosis but is important to highlight the chronicity of this disease. Notable findings on biopsy include pikiloderma – telangiectatic skin lesions associated with chronic inflammation, often in sun-exposed areas [3]. Like dermatomyositis, it is associated with malignancy (4). The diagnosis is supported by the presence of anti-MDA-5 antibodies [4],[5] , but not definitively confirmed.Notably, ILD is near universal in ADM. There is also significant clinical and serologic overlap between ADM and other autoimmune conditions [6],[7]. | ||||||
|
CASE REPORT
| ||||||
|
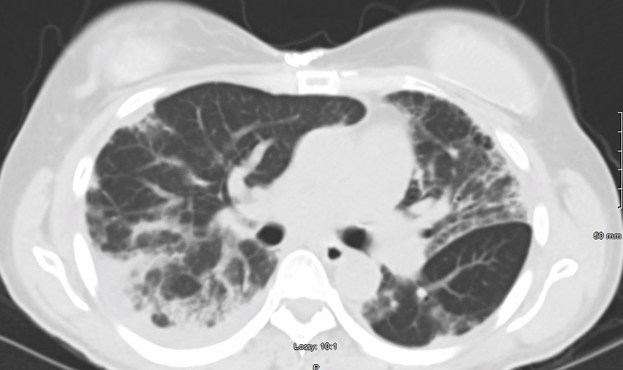
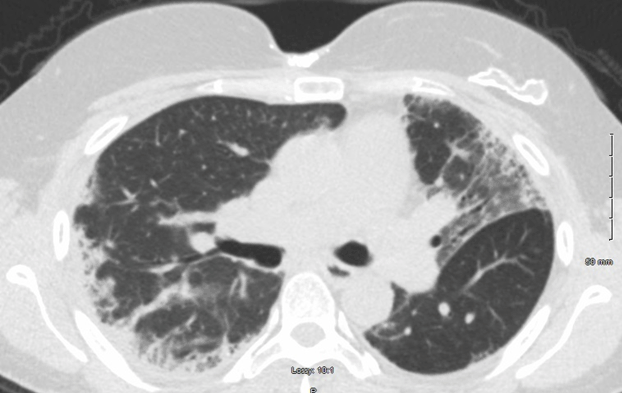
A 44-year-old Hispanic female with a past medical history of Hypothyroidism presented from her endocrinologist with a diffuse, maculopapular rash, including the face, hands, wrists, torso, arms and legs. The rash had been intermittent for the past six years, painful, pruritic, and exacerbated by sunlight. She also complained of joint pain and swelling, myalgia and exertional dyspnea. Physical exam revealed hyperpigmentation of the metacarpophalangeal joints (Figure 1) and diffuse joint swelling. However, muscle strength was mostly intact. Examination of her face showed alopecia, periorbital swelling and erythema. Pulmonary auscultation revealed fine crackles. Dermatomyositis, complicated by an underlying connective tissue disease, leading to ILD was considered. Initial serology was significant for high titers of anti-nuclear antibodies with cytoplasmic staining, alongside smooth-muscle and anti-cardiolipin antibodies. Computed Tomography confirmed interstitial lung disease with fibrosis. However, her CPK and aldolase levels were within normal limits, suggesting an Amyopathic Dermatomyositis. Given the normal CPK and Aldolase levels, the patient was spared a muscle biopsy. Serology for Anti-MDA-5 antibodies was unavailable due to rarity. Accordingly, treatment was started based on the clinical picture alone. She failed two months of first-line treatment with Azathioprine: initially dosed at 50mg twice daily for one month, and then increased to thrice daily for a month. When she didn’t improve, she was switched to cyclophosphamide, 750 mg/m2 IV, once monthly, for a total of 6 months. Simultaneously, she was started on mycophenolate 750 mg twice daily, and titrated down to 500mg twice daily based on her symptoms. She improved dramatically within the first month of cyclophosphamide and mycophenolate combination therapy. With regards to her pulmonary fibrosis, she was started on Nintedanib 150mg twice daily. Her dyspnea improved, and she was tapered down to 100 mg twice daily. Clinical improvement was mirrored by radiological improvement on computed tomography taken at diagnosis and two years later (Figures 2 and 3). She has been maintained on Mycophenolate 500 mg twice daily and Nintedanib 100mg twice daily for over two years without significant relapse. Malignancy screening including computed tomography of the chest, mammogram, pap smear, and colonoscopy were negative. | ||||||
| ||||||
| ||||||
|
| ||||||
DISCUSSION | ||||||
|
ADM is distinguished as the cutaneous findings of dermatomyositis, but without the signs or symptoms of myopathy [1]. Signs usually include Gottron’s papules: violaceous pigmentations on the dorsal aspects of the knuckles, and ulcerations of the elbows (Gottron’s sign), digital pulp and nail beds [2]. Patients may also present with oral ulcers, arthritis and palmar macules [8]. Like DM, ADM patients are at an increased risk of malignancy. ADM is more commonly associated with breast, ovarian, and nasopharyngeal cancers compared to DM which is more commonly associated with lung cancer [4]. Accordingly, patients should be monitored and screened with colonoscopies, pap smears and mammograms. Patients with ADM are at an increased risk of pulmonary fibrosis, and carry a risk for rapidly progressive lung disease, which often proves fatal [9]. There is significant overlap between connective tissue diseases such as SLE, Scleroderma, Mixed Connective Tissue Disease and less often, Rheumatoid Arthritis and Sjogren’s Syndrome [6], [7]. This overlap was evident in our patient. She had aspects of lupus and auto immune hepatitis, with clinical photosensitivity and serological positivity for ANA, cardiolipin and smooth-muscle antibodies.
Epidemiology
MDA-5 antibody There are no good clinical trials for ADM, and first-line treatment protocols are still being explored [12]. Given the cutaneous manifestations of ADM, many physicians have focused on the treatments for the cutaneous aspects of DM. However, this has proved ineffective. One review found a majority of patients received hydroxychloroquine, but the majority of them (55%) did not respond and required escalated care in the form of higher dosed steroids, cyclophosphamide, azathioprine, or mycophenolate [12]. The presence of pulmonary fibrosis is known tobe a poor prognostic indicator [13]. The danger is that mild fibrosis may develop into rapidly progressive interstitial lung disease, which is often treatment refractory and quickly fatal [13]. Accordingly, faster escalation of care is required. Specific treatment guidelines are not yet available. Some recent cases have documented the success of tacrolimus [14] and intravenous immunoglobulin (IVIG) [12] when Mycophenolate or Azathioprine have failed. Rituximab has also been shown to be successful, and can be used according to the Rituximab in Myositis (RIM) study [15]. In general, treatment of more severe cases seems to follow that of other myositic conditions. Begin therapy with high dose steroids, followed quickly by steroid sparing agents in an attempt to taper the dose. Given the presence of pulmonary fibrosis in our patient, treatment of the cutaneous disease alone was insufficient. She was started on high dose steroids but failed azathioprine as a steroid sparing agent. She responded well to cyclophosphamide induction, and mycophenolate maintenance. Her pulmonary fibrosis was caught early, and has been well controlled on mycophenolate with the addition of Nintedanib. The patient reported significant clinical improvement in her dyspnea and cutaneous manifestations. The patient did not receive IVIG nor respond to azathioprine, but improved on cyclophosphamide and mycophenolate. Notably, the diagnosis was made without MDA-5 serology, and treatment was rendered based on the clinical picture and labs. The presence of anti-cardiolipin, anti-nuclear and smooth muscle antibodies highlights the overlap with other autoimmune conditions and the difficulty in diagnosing this potentially fatal disease. | ||||||
|
CONCLUSION
| ||||||
|
Due to the association with malignancy and the higher incidence of pulmonary fibrosis and rapid disease progression in ADM compared with other myositic conditions, early detection and treatment is vital. Treatment is debated, but usually revolves around immune-suppressants and steroid sparing agents, along with IVIG or Rituximab if needed. | ||||||
|
REFERENCES
| ||||||
| ||||||


|
[HTML Abstract]
[PDF Full Text]
|
|
Funding
Funding, where applicable, was split between the authors and the home institution. |
|
Patient Protections
Given that this is a case report, without any personally identifying information, informed consent was not required. All research was conducted with respect for persons, in accordance with the Declaration of Helsinki. |
|
Author Contributions
Luis W. Dominguez – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Marissa Sansone – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published Valentin Marian – Substantial contributions to conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Revising it critically for important intellectual content, Final approval of the version to be published |
|
Guarantor of Submission
The corresponding author is the guarantor of submission. |
|
Source of Support
None |
|
Consent Statement
Written informed consent was obtained from the patient for publication of this case report. |
|
Conflict of Interest
Author declares no conflict of interest. |
|
Copyright
© 2018 Luis W. Dominguez et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|